杉野研究室

研究テーマ
- 非平衡統計力学的アプローチによる物質中の電子移動反応の理解
- 物質中の水素・ミュオンの量子状態
- 電子格子相互作用の第一原理計算
- 超伝導体の第一原理計算
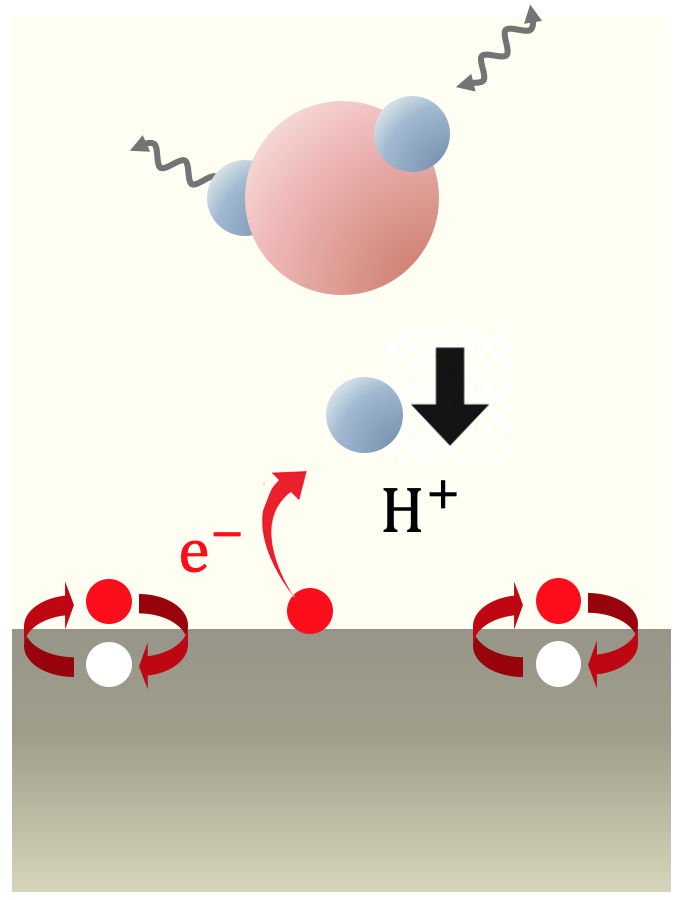
電子とイオンが連動して起こる化学反応によるエネルギー変換過程を、非平衡運動論の枠組みで捉えるための研究を行っている。今回は、プロトンが電極水溶液界面において電子およびフォノンを励起しながら運動エネルギーを失って表面に吸着するVolmer過程を、非平衡グリーン関数法の枠組みで探った。本計算は、先行研究を拡張してプロトンの運動をあらわに考慮したものになっている。従来格子摩擦が支配的だと考えられてきたが、電極での電子正孔対を励起することで起こる電子摩擦が同様に重要であることが分かった。燃料電池等で起こるエネルギー変換を捉え直す必要性を示唆する結果である。目下、第一原理計算に基づいて現実系により忠実に反映した模型を用いた研究を行っている。