Ozaki Group

- Affiliation
-
Numerical Materials Research Laboratory (NML)
(concurrent with Division of Condensed Matter Theory, Division of Data-Integrated Materials Science) - Course
- Phys., Sci.
 Research Associate FUKUDA, Masahiro
Research Associate FUKUDA, Masahiro
Research Subjects
- Development of efficient and accurate methods for first-principles electronic structure calculations
- Development of the OpenMX software package
- Development of first-principles methods for X-ray spectroscopies
- First-principles calculations of surfaces and two-dimensional structures
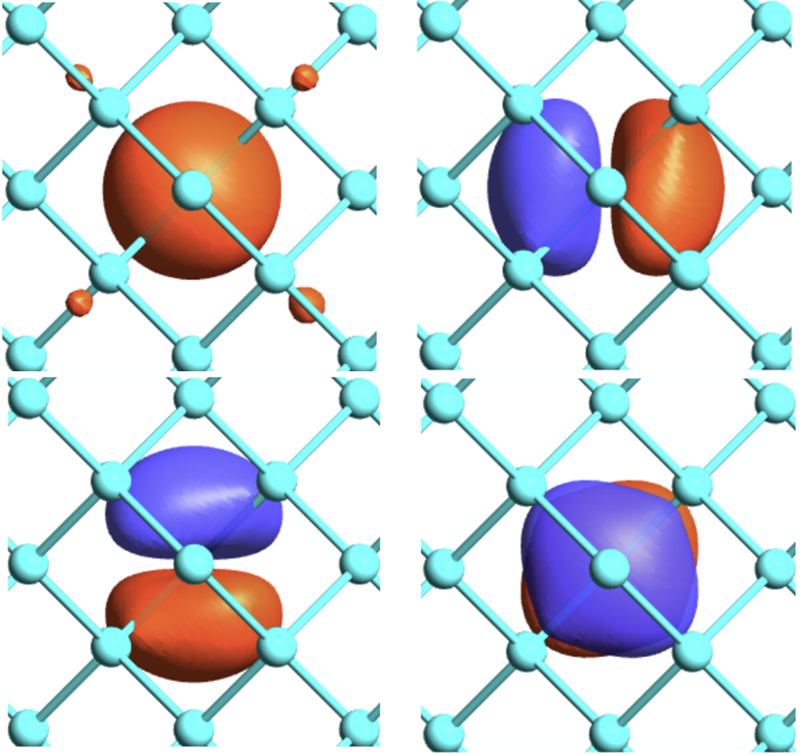
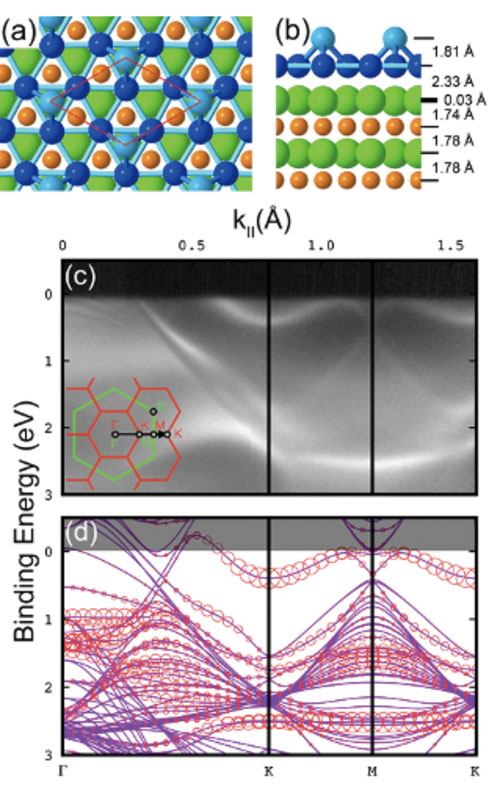
With the development of massively parallel computers and the increasing demand for precision in materials science, first-principles electronic structure calculations have become increasingly important, not only for understanding individual materials but also as a foundation of computational materials science. Based on density functional theory, we pursue both the development of computational methods for describing complex real materials with high reliability and the advancement of OpenMX as a core software platform. Using a versatile atomic-orbital basis-set scheme, we have developed original methodologies rooted in both mathematical insight and computer science, including the order-N divide-and-conquer method based on localized natural orbitals, a domain-decomposition method based on the modified recursive bisection approach, and a parallel fast Fourier transform method designed to minimize communication. In recent years, we have been working toward the further development of density functional theory through studies on absolute core-electron binding energies, the nearest Wannier function method, and exchange-correlation functionals. We also study a wide variety of systems, including material surfaces and two-dimensional materials, in close collaboration with experimental groups, with the aim of elucidating the relationship between structure and electronic states while extending the applicability of first-principles calculations to real materials.