
Carrier Transport in Gradient-Doped Photoelectrochemical Electrodes
Lippmaa Group
The interest in developing electrode materials for photoelectrochemical water splitting cells stems from a vision of a sustainable and pollution-free hydrogen-based energy cycle. In essence, the technique is reliant on the development of wide-gap semiconductor materials that are electrochemically and structurally stable over an extended time period, measured in years, in a hydrogen or oxygen evolution reaction under sunlight illumination. Perovskite titanates such as SrTiO3 provide the prerequisite stability in water, but suffer from a variety of intrinsic factors that rate-limit the solar-to-chemical energy conversion process. The primary rate-limiting issues are the non-optimal band gap of 3.2 eV and a short, sub nanosecond photocarrier lifetime. The band gap can be brought down closer to the optimal value of around 2 eV by doping, but adding dopants to the crystal also tends to further shorten the carrier lifetime to picosecond range, negating any possibility of high-efficiency diffusive photocarrier transport from the bulk of the semiconductor to the water interface to drive the water splitting reaction.
We have recently attempted to determine the role of Ti-site dopants on the energy conversion efficiency under ultraviolet band-gap excitation using non-doped and Nb-doped SrTiO3 single crystals (A-D) shown in Fig. 1. The corresponding cyclic voltammetry curves are shown in Fig. 2, where the current density is directly proportional to the efficiency of the energy conversion process, i.e., the number of generated gas molecules. The photoresponse of a non-doped 0.5-mm-thick SrTiO3 crystal (A) is zero due to the large resistance of the insulating crystal, which prevents current flow in an electrochemical cell (PEC) illustrated in Fig. 1(A-D). Photoinduced holes in the surface depletion layer of the substrate crystal, marked with a yellow gradient in the PEC diagram in Fig. 1, drive the oxygen evolution reaction but the short lifetime of photocarriers prevents diffusive photoelectron transport through the bulk part of the crystal, stopping the water splitting reaction.

Fig. 1. (A to D) Nb:SrTiO3 single-crystal electrode materials, with doping levels of A:0%, B:1%, C:0.1%, D:0.04%. (A-D) photoelectrochemical cell configuration. Electrode crystal shown in violet, surface depletion layer in yellow. (E,F) Cell configuration for thin film samples consisting of a SrTiO3 film (yellow) on a metallic Nb:SrTiO3 substrate (violet).
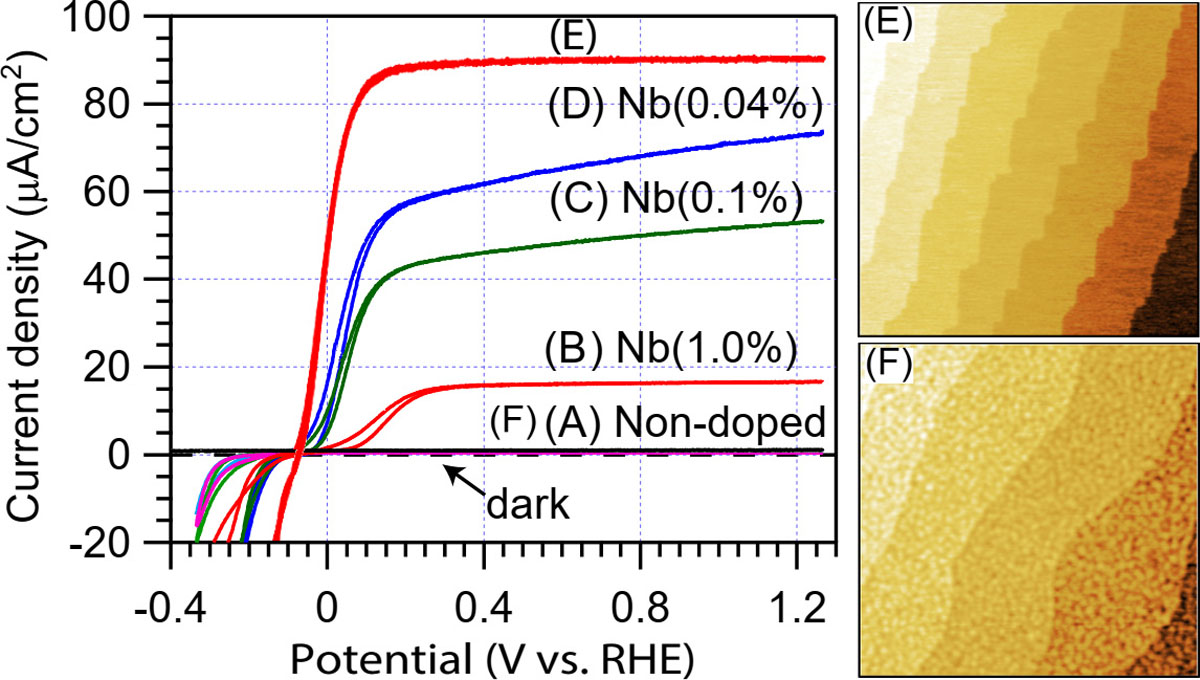
Fig. 2. Illustration of energy conversion efficiency, which is proportional to the cyclic voltammetry current for crystals A to F. The atomic force microscope images for films E and F illustrate the difference in surface morphology related to lower (E) and higher (F) point defect densities in the films.
Electron transport becomes possible when bulk conductivity is induced by slight Nb doping of SrTiO3 at the Ti site. As shown in Fig. 2, the efficiency gradually increases as the doping level is reduced from 1% to 0.04% (B to D). Further doping reduction will lead to reduced efficiency due to increasing internal resistance of the substrate crystals.
Improvement of the PEC efficiency can be achieved with a bilayer heterostructure design that combines a non-doped defect-free SrTiO3 crystal in the ~100 nm thick surface depletion region of the electrode with a slightly-doped Nb:SrTiO3 substrate, illustrated in Fig. 1(E,F). Photocarrier recombination losses are minimized and extraction efficiency maximized in the depletion region where the high internal field associated with the surface band bending drives fast carrier transport, whereas bulk doping is used to promote electron transport in the bulk diffusive region of the electrode. Ideally, a continuously varying doping gradient could be used to match the bulk doping level to the width of the surface depletion layer by growing a crystal with a depth-direction gradient of the doping level.
As a simplified demonstration of this concept, we grew a high-crystallinity film with a minimal point defect density and an atomically flat surface morphology (Fig. 2, E) on a highly-doped Nb:SrTiO3 substrate [1]. The importance of defect density control can be seen in a comparison electrode, which was grown at a low temperature and thus contained a high density of point defects (Fig. 2, F). The energy conversion performance of the defect-rich film was, as expected, nearly zero.
This work demonstrates the importance of point defect density control in PEC electrode materials and shows that depth-graded doping control can be a viable strategy for designing efficient hydrogen generation photoelectrochemical electrodes.
References
- [1] S. Kawasaki, R. Takahashi, and M. Lippmaa, J. Phys. Chem. C, 123, 15551 (2019).