
Adsorption and Reaction of Formic Acid on Cu(111)
PI of Joint-use project: I. HamadaHost lab: Yoshinobu Group
Hydrogen is one of the promising energy sources toward the carbon neutral and sustainable society. Formic acid (HCOOH) has attracted attention as a candidate for the hydrogen storage material because it is nontoxic and exists as a stable molecule at standard temperature and pressure conditions. In general, HCOOH can be catalytically converted into CO2 and H2 via dehydrogenation or into CO and H2O via dehydration. Among others, a Cu surface is one of the attractive catalysts, because HCOOH is selectively decomposed into CO2 and H2 via the formate (HCOO) intermediate without generating CO. However, the mechanism of HCOOH dehydrogenation is not yet fully understand. One of the reasons may be that the state of HCOOH on a Cu surface, in which HCOOH is adsorbed in a polymeric form, is not yet determined. Furthermore, there is a discrepancy between the computational and experimental adsorption energies [1]. Toward the full understanding of the HCOOH dehydrogenation on Cu catalyst, we conducted density functional theory studies on monomeric as well as polymeric adsorption of HCOOH on Cu(111), as well as their dehydrogenation reactions.
We used the rev-vdW-DF2 [2], a variant of the van der Waals density functional (vdW-DF). Use of vdW-DF is essential, as the interaction between HCOOH and Cu surface is very weak and is of dispersion type. The surfaces were represented using slab models, and the electrostatic interaction in the surface normal direction was treated rigorously by using the effective screening medium method.
We first investigated the monomeric adsorption of HCOOH on Cu(111) and found that the adsorption energy is much smaller than the experimental value, when the generalized gradient approximation is used, while it is in better agreement with the experiment, when the dispersion forces are properly described via vdW-DF [3].
We then investigated the molecular structures for the polymeric HCOOH adsorption. In the literature, there have been proposals of the so-called α- and β-polymeric structures on the surfaces (Fig. 1), according to the crystalline HCOOH, but the stable form on Cu(111) has yet to be resolved. We explored the stable structure of polymeric HCOOH by considering various structures and in-plane periodicities. HCOOH interacts weakly with the surface, while it interacts with the neighboring molecules via the H-bonding-like interaction. We found that the β-polymeric form is energetically more stable than the α one, although they are energetically competing. However, by simulating the vibrational spectra, scanning tunneling microscopy/atomic force microscopy (AFM) images (Fig. 2), and by comparing with the experiments, we concluded that α-polymeric HCOOH is more plausible and has been formed and observed in the experiments [4, 5]. We then studied the dehydrogenation reaction by taking the edge effect into account. We found that the dehydrogenation reaction is facilitated at the edge of the polymeric HCOOH chain, and it barely occurs within the polymeric HCOOH chain. Our results are consistent with the previous experiments.
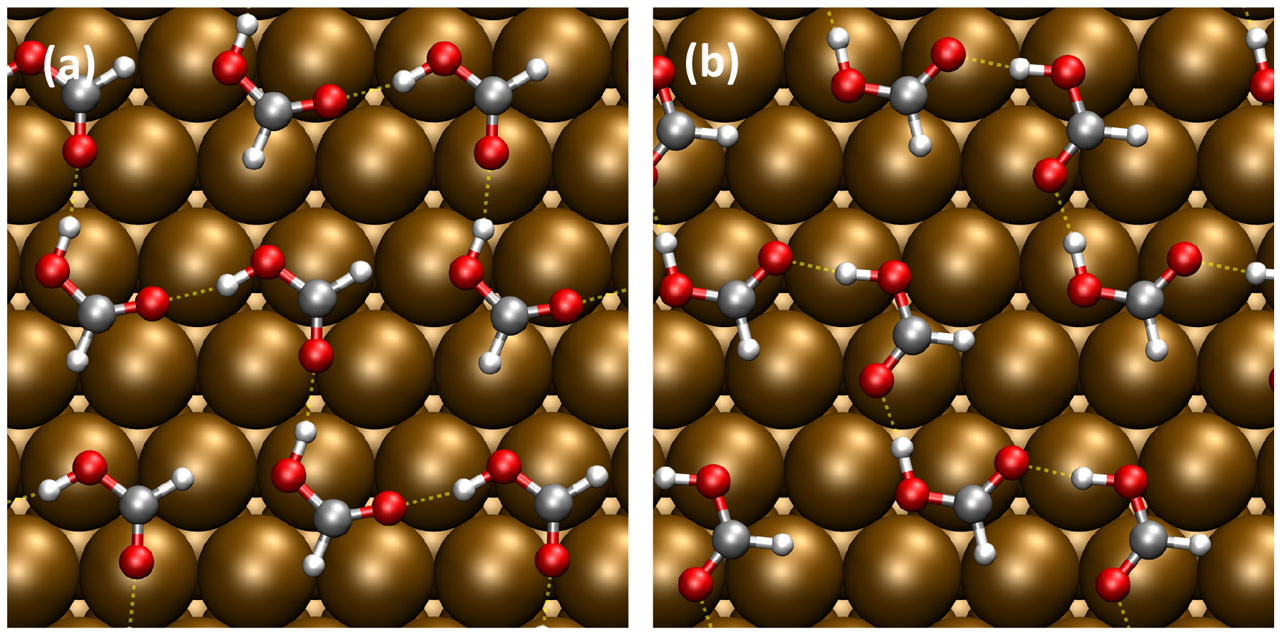
Fig. 1. Structures of (a) α-polymeric and (b) β-polymeric HCOOH’s on Cu(111).
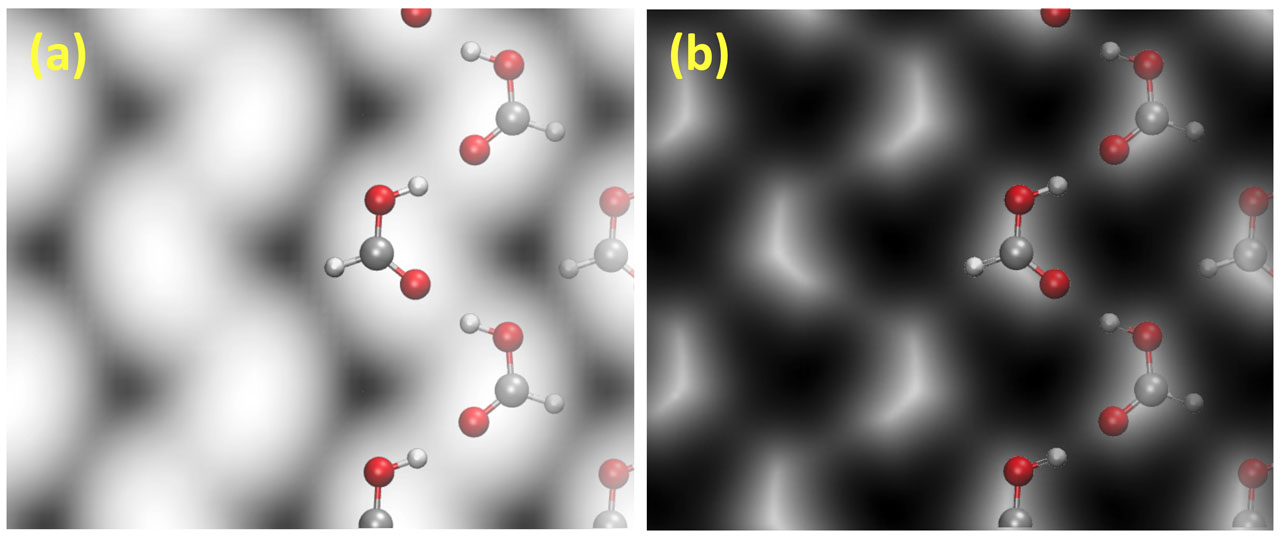
Fig. 2. (a) Simulated scanning tunneling microscopy image and (b) simulated atomic force microscopy image of polymeric HCOOH in the α-form on Cu(111). Tersoff-Hamann and probe particle methods were used to generate the former and the latter, respectively.
The polymeric HCOOH is decomposed (in part) into HCOO and form complex nanostructures, depending on the annealing temperature. We performed extensive structure search of HCOOH-HCOO and HCOO with different compositions and successfully determined the structures of chain-like monodentate HCOO + HCOOH and monodentate HCOO + bidentate HCOO aggregates as well as bidentate HCOO clusters by evaluating the adsorption energies and simulating the AFM images [4].
In summary, we have clarified the adsorption states of HCOOH as well as the aggregates composed of HCOOH and HCOO and the mechanism of the dehydrogenation of HCOOH into HCOO on Cu(111), and succeeded in tracking the fate of HCOOH on Cu(111) upon annealing.
References
- [1] Y. Shiozawa, T. Koitaya, K. Mukai, S. Yoshimoto, and J. Yoshinobu, J. Chem. Phys. 143, 234707 (2015).
- [2] I. Hamada, Phys. Rev. B 89, 121103(R) (2014).
- [3] S. E. M. Putra, F. Muttaqien, Y. Hamamoto, K. Inagaki, I. Hamada, and Y. Morikawa, J. Chem. Phys. 150, 154707 (2019).
- [4] A. Shiotari*, S. E. M. Putra*, Y. Shiozawa, Y. Hamamoto, K. Inagaki, Y. Morikawa, Y. Sugimoto, J. Yoshinobu, and I. Hamada, Small 17, 2008010 (2021). (*: equal contribution)
- [5] S. E. M. Putra, F. Muttaqien, Y. Hamamoto, K. Inagaki, A. Shiotari, J. Yoshinobu, Y. Morikawa, and I. Hamada, Phys. Rev. Materials 5, 075801 (2021).