
Advanced First-Principles Simulation of Electrochemical Interfaces
S. Kasamatsu, I. Hamada, and O. Sugino
Electrochemical interfaces have long been attracting attention as an environment for the interconversion of chemical and electric energies, but the first-principles simulation is not yet sufficiently matured because of the difficulties in (i) achieving the accuracy required for the reliable prediction and (ii) modeling the electric double layers within the density functional theory (DFT). As a step for overcoming, we studied accuracy of the random phase approximation (RPA) for the exchange and correlation (xc) of electrons and then developed an ab initio Monte Carlo method suitable for sampling the distribution of defects in an electrode.
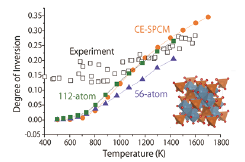
Fig. 1. Plot of the degree of conversion versus the temperature (T) and the MgAl2O4 spinel structure (inset). Our calculation using 112-atom and 56-atom cells well reproduced the previous simulation based on a cluster expansion model (CE-SPCM) in the whole range of T and also the neutron experiment at higher T. First-principles sampling for solids has thus made feasible.
Hydrogen (H) on a platinum (Pt) surface, H/Pt(111), is a typical benchmarking system for which many theoretical and experimental studies exist, but controversies still remain even for the H adsorption problem: H is presumed to adsorb on the fcc site based on indirect experiments and the conventional DFT simulations, while there exist several reports that signal from the top site can be observed spectroscopically. Since the conventional DFT, which is based on the semilocal approximation for the xc potential, was shown inaccurate for the sorption problem on Pt(111), we have tried to use more advanced xc potential called RPA [1]. The result of the calculation accurately reproduced the experimental adsorption energy although conventional DFT considerably overestimates it. Contrary to the conventional DFT, the DFT-RPA predicts that difference in the energy between the fcc and top sites is on the order of thermal energy at room temperature, i.e. kBTroom, suggesting coexistence of the fcc and atop hydrogens. This renewed picture on the H adsorption explains why signal from the top site may be observed. We believe this is an important step for true microscopic understanding of the hydrogen evolution reaction, which is one of the most important fuel-cell reactions.
The fuel-cell reaction occurs efficiently on the Pt electrode but the efficiency needs further improvement to meet the demand of future technology. In this context, oxide electrode (TiO2 or ZrO2) has attracted considerable attention as a beyond Pt material. By introducing impurities and oxygen vacancies, the oxides are made active for the fuel-cell reactions. Theoretically, however, it has been extremely difficult to model this system by sampling the distribution of defects for appropriate description of the space charge layer; this is because of the large barrier existing between different configurations of defects. The ab initio molecular dynamics (MD) simulation is an established method for the sampling but is generally inadequate for solids. In this context, we have developed an ab initio Monte Carlo (MC) method of replica exchange type, which runs very efficiently on massively parallel supercomputers. Our benchmark calculation on MgAl2O4 spinel shows that the degree of inversion, or the ratio of Al ions on Mg sites, is successfully sampled in the temperature range studied [2] (Fig. 1). This is considered as an important step for elucidation of the reason why the oxides so effectively activate the fuel-cell reactions.
References
- [1] L. Yan, Y. Sun, Y. Yamamoto, S. Kasamatsu, I. Hamada, and O. Sugino, J. Chem. Phys. 149, 164702 (2018).
- [2] S. Kasamatsu and O. Sugino, J. Phys.: Condensed Matter, 31, 085901 (2019).