
Efficient Method for Membrane Protein Structure Predictions
Y. Okamoto
Membrane proteins have essential biological functions based on their structures for life, and atomistic structural information is important to investigate the mechanism of their functions. Computers have been developing for decades. However, the resource runs short of structure predictions by brute-force simulations yet. Thus, efficient algorithm for computer simulation is needed to reduce computational resources for the prediction.
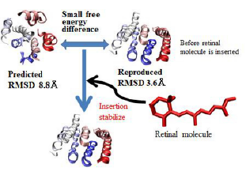
Fig. 1. Two predicted structures from our simulation and hypothesis about the relation between the structures and effects of retinal molecule insertion for the structures. In other words, the hypothesis is as follows: from our simulation without retinal molecule, the difference between above two structures is not large, however, insertion of retinal molecule into the empty space results in the stabilization of native-like structure compared to the other structure. This hypothesis is also consistent with previous experimental results.
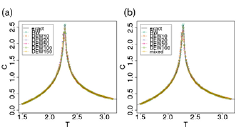
Fig. 2. Specific heat of 2-dimensional Ising model obtained by conventional random walk REM, DEWREM, and mixed simulations with (a) Metropolis replica-exchange method and (b) DETREM. The error bars are smaller than the symbols. In the inset, the labels are as follows. exact: exact solution, RW: random walk, DEWn: DEWREM with the interval of n MC steps between replica exchanges, and mixed: mixed walk.
There are two fundamental approaches for the developments: decrease of interaction points in system and enhancement of sampling. A former example is to adopt an implicit solvent model. In good models, this method can achieve the structure prediction with simulations. A latter example is to employ replica- exchange method (REM) which enhances sampling of conformations. This method typically accelerates the crossing of free energy barriers using temperature change of replicas.
We developed the method from both approaches. We extended the previous method to target most membrane proteins [1]. Using the method, we performed structure prediction of bacteriorhodopsin and reproduced the native structure. Moreover, we also obtained the expected but unknown structure with empty space structure for retinal insertion, which is related to a hypothesis for co-molecule insertion during membrane protein folding as is shown in Fig. 1. As a result, our simulation produced the consistent results for previous experiments and supported the membrane protein folding hypothesis in co-molecule stage. Because we confirmed the validity of our new method for structure predictions, a next step will be to predict the unknown membrane protein structures or low-resolution protein structures.
We next proposed two new replica-exchange methods to increase efficiency of REM. One is the method of replica exchange with a differential equation without pseudo random numbers [2]. The other is the method that specifies the order of replica exchange and, thus, the trajectory in temperature space among replicas [3]. With 2-dimensional Ising model, we compared results of these new methods and its combination to results of the conventional Metropolis replica-exchange method. Figure 2 shows that specific heat obtained from the conventional Metropolis replica-exchange method (METREM), deterministic replica-exchange method (DETREM), designed-walk replica-exchange method (DEWREM), and their combination. DETREM with random-walk replica-exchange method reproduced the exact quantities including phase transition. Because short interval causes correlation among replicas, DEWREM requires a longer interval of replica- exchange attempts to reproduce the conventional results. On the other hand, biomolecular simulation generally employs longer time interval between replica-exchange attempts. Hence, this does not cause such a problem in such simulations. In these simulations, DEWREM will result in the increase of efficiency of conformational sampling. For spin system, another implementation such as mixed walk simulation or different pairs in designed walk can improve the interval problem. These are future works to improve these methods.
References
- [1] R. Urano, H. Kokubo, and Y. Okamoto, J. Phys. Soc. Jpn., in press.
- [2] R. Urano and Y. Okamoto, arXiv:1412.6959.
- [3] R. Urano and Y. Okamoto, arXiv:1501.00772.