
Accurate Prediction of Chemical Reaction Systems
Sugino Group
The functional material research, one of the priority subjects of ISSP, investigates the link between (a) various functions of a heterogeneous system and (b) the basic properties of electrons and nuclei. The first-principles simulation can contribute to the research by, for example, (i) obtaining the potential energy surface (PES) for the nuclei and (ii) simulating the non-equilibrium chemical reaction dynamics occurring thereon. Sugino group has been working on the latter subject (ii) by developing algorithms such as the effective screening medium (ESM) [1] for applying the bias potential to the system and the potentiostat molecular dynamics scheme [2] for controlling the bias, with which to study the electrode-electrolyte interface problems. On the other hand, methods for the former subject (i), or obtaining reliable PES, has been left unsolved although crucially important for the present purpose.
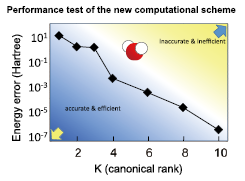
Fig. 1. A version of the extended symmetric tensor decomposition (ESTD) scheme applied to the water molecule. The total energy of the water referred to the full configuration interaction (FCI) is plotted against the canonical rank (K). The total energy converges exponentially to the FCI indicating remarkable efficiency of the computational scheme, which is well beyond most of the conventional schemes.
Our study in the latter direction began by reformulating the full configuration interaction (FCI) method, or the exact diagonalization method, which is formally exact within the chosen basis set but the application is severely restricted by the combinatorial explosion of the degrees of freedom. It has been occasionally suggested that the key to reducing the degrees of freedom is in a construction of a specialized molecular orbital (MO) set that is non-orthonormal and spatially localized. With this in mind, an efficient and robust algorithm was developed [3, 4], wherein the many-body electronic wavefuntion (WF) is described using a canonical tensor format and the MOs are variationally determined so as to achieve the most compact representation of WF. In the most recent version [4], the resulting WF has a form of a linear combination of the antisymmetrized geminal powers (AGPs), where AGP is equivalent to the Hartree-Fock-Bogoliubov (HFB) state with a fixed particle number. This means that the computational scheme developed herein describes the interacting geminals while the conventional FCI describes the interacting unpaired electrons. The total energy formulas were derived originally for this purpose but many of them are found equivalent to those used in the nuclear physics in a different context.
When the new scheme, called extended symmetric tensor decomposition (ESTD), is applied to, for example, the water molecule that is a benchmark ten-electron system, the WF is accurately described only with ten terms for the decomposition series, i.e., ten for the canonical rank (K) (Fig. 1). The computational time to obtain the total energy within μHartree error scales as O(K2M5), where M is the number of MOs that is proportional to the number of electrons, of which the scaling is superior to most of the existing computational schemes. This remarkable result indicates a promise of this scheme in improving the accuracy of PES within reasonable computational time. This scheme will allow further improvements: when a more elaborate tensor format is used instead of the canonical one, one would achieve further accuracy taking smaller rank; when a mean-field approximation is combined to ESTD to simplify the long ranged correlation that will be weak in some chemical reaction systems, one would achieve further efficiency. The ESTD scheme is general and can be adapted to such modifications. We thus expect ESTD will finally overcome known inaccuracy of the density functional PES and will prove a powerful tool not only to the chemical reaction systems but also many other systems.
References
- [1] M. Otani and O. Sugino, Phys. Rev. B 73, 115407 (2006).
- [2] N. Bonnet, T. Morishita, O. Sugino, and M. Otani, Phys. Rev. Lett. 109, 266101 (2012).
- [3] W. Uemura and O. Sugino, Phys. Rev. Lett. 109, 253001 (2012).
- [4] W. Uemura, S. Kasamatsu, and O. Sugino, arXiv:1504.06250v1 (2015); to be published in Phys. Rev. A.