Sugino Group

- Affiliation
-
Functional Materials Group
(concurrent with Division of Condensed Matter Theory, Materials Design and Characterization Laboratory, Center of Computational Materials Science) - Course
- Phys., Sci.
Research Subjects
- Understanding electron transfer reactions in materials using a non-equilibrium statistical mechanics approach
- Quantum states of hydrogen and muon in a material
- Electron-phonon couplings from first principles
- First-principles simulation of superconductors
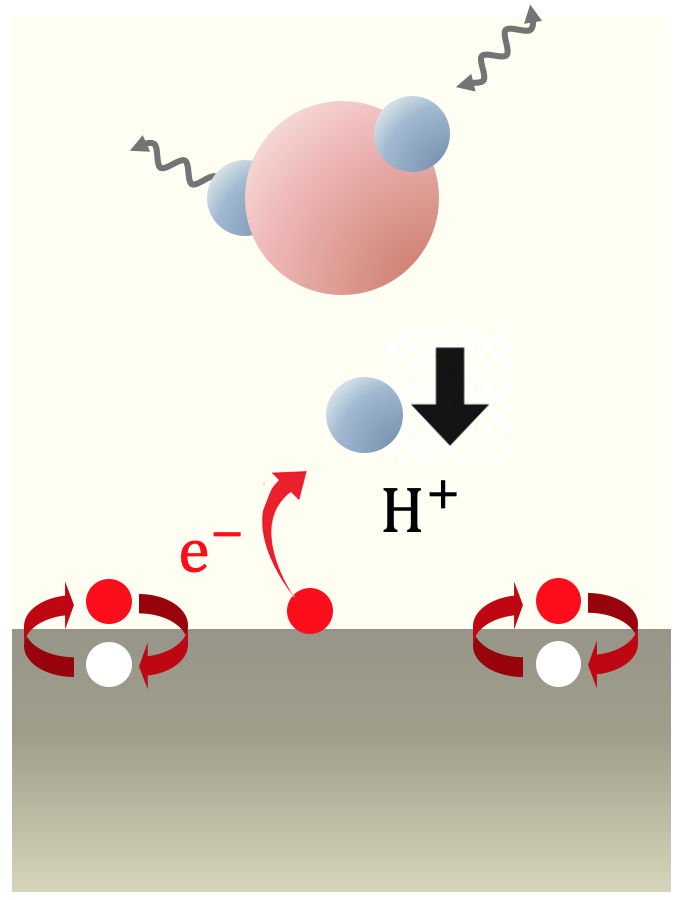
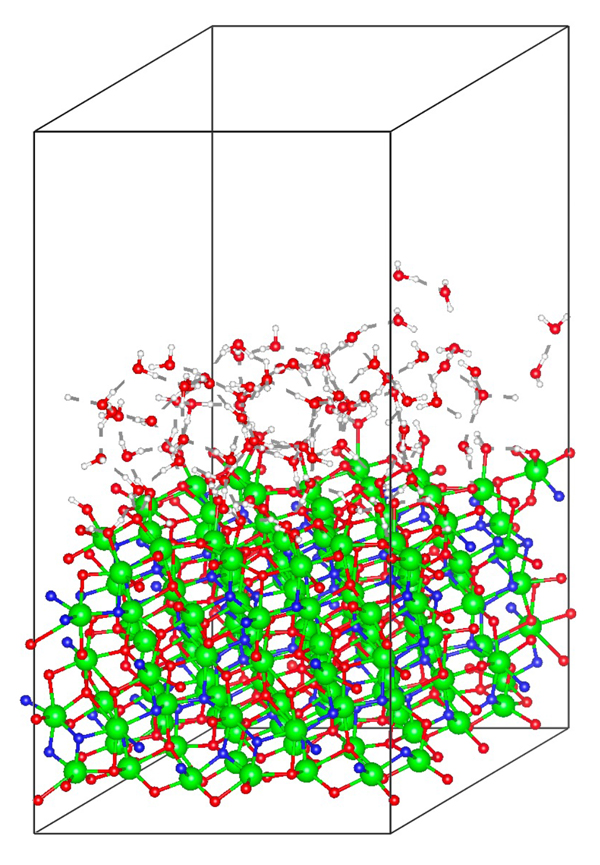
This study explores energy conversion processes driven by chemical reactions involving electrons and ions within the framework of non-equilibrium kinetics. Specifically, the Volmer process, where protons lose kinetic energy and adsorb onto the surface while exciting electrons and phonons at the electrode-solution interface, was examined using the non-equilibrium Green's function method. This calculation extends previous research by explicitly considering proton motion. Traditionally, lattice friction has been considered dominant, but our findings reveal that electron friction, caused by the excitation of electron-hole pairs at the electrode, is equally significant. These results suggest the need to reconsider energy conversion processes occurring in fuel cells. Currently, research is being conducted using models that more faithfully reflect real systems based on first-principles calculations. This approach aims to provide a more accurate understanding of the mechanisms involved in energy conversion processes, potentially leading to more efficient fuel cell designs and other applications in the field of energy technology.