
Material Design of Luminescence from First-Principles
Sugino Group
Many-body Green’s function methods have attracted considerable attention as emerging computational methods to allow prediction of excited states of a material from first-principles. Breakthrough was achieved by the algorithms developed for massively parallel supercomputers and by the approximations made for balancing the computational cost and accuracy. The software package developed by this group, for example, is now able to handle up to 200 atoms in a cell, sufficiently large for many applications, and provide in addition the wave function of the electron-hole pair (exciton) containing rich information of the excited states. The utilization of the exciton wave function, however, has been done infrequently, and in this context, this group proposed a method to obtain from the wave function a number of quantities that can characterize the excitons. The obtained quantities are found particularly useful in classifying the excitons into the local, charge-transfer (CT), Rydberg excitons [1], so that one may predict the luminescence dynamics induced by photoabsorption. This novel method opens possibility of a computational material design toward efficient luminescence.
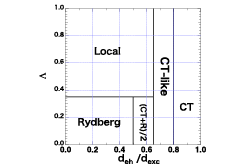
Fig. 1. Λ- deh/dexc map proposed in Ref. [1] to classify excitons into local, charge-transfer (CT), Rydberg, and others such as CT-like excitons. This map has enabled to relate the exciton wave function with the characteristics of excitons, and thus relate the characteristics with the structure of a material, making it significantly easier to computationally design a luminescent material.
The exciton wave function is a six-dimensional quantity, which has been considered to carry too prodigious amounts of information. With increasing ability of modern supercomputers, however, it has become easy to use the wave function to obtain various quantities; for example, one can calculate the exciton binding energy by taking expectation value of the electron-hole interaction operator, the electron-hole separation deh from the distance between the electron and hole, and the exciton size from the square root of the distance. When using (a) a quantity characterizing the range of the exciton Λ and (b) the electron-hole separation distance deh relative to the exciton size dexc, one can plot each excited state in a two-dimensional Λ- deh/dexc map (Fig. 1), wherein excitons with different type are let distribute in a different region. This method was proven useful in our subsequent study on the photoabsorption spectra of the recently synthesized cycloparaphenylene molecules [2] and on the singlet-triplet splitting of the thermally activated delayed fluorescence (TADF) molecules [3].
References
- [1] D. Hirose, Y. Noguchi, and O. Sugino, J. Chem. Phys. 146, 044303 (2017).
- [2] Y. Noguchi and O. Sugino, J. Chem. Phys. 146, 144304 (2017).
- [3] Y. Noguchi and O. Sugino submitted to J. Phys. Chem.