
Capturing Transiently Charged States at the C60/TiO2(110) Interface by Time-Resolved Soft X-ray Photoelectron Spectroscopy
K. Ozawa, S. Yamamoto, and I. Matsuda
One of key technologies for realizing sustainable society is an efficient conversion of the sunlight into chemical and electrical energies. Titanium dioxide (TiO2) exhibits a high photocatalytic activity for water splitting to produce a zero-emission fuel. TiO2 is also used as a transparent electrode in photovoltaic cells [2]. One of the extensively studied TiO2-based photovoltaic cells is a dye-sensitized organic cell, in which visible-light sensitive dye molecules or polymers are loaded onto the TiO2 electrodes. The TiO2 electrode collects photoexcited electrons from the light absorbing layer to facilitate electron-hole separation. In such a photovoltaic cell, a charge transfer across the interface is an important elemental step to define the performance of the cell. A molecule of C60 is an important component of dye-sensitized photovoltaic cells, where the C60 layer is used as an electron acceptor and as an electron transport layer. C60 also serves to enhance photocatalytic activity of TiO2 by promoting the separation of the photoexcited electron-hole pairs. In the present study, we apply time-resolved soft X-ray photoelectron spectroscopy (TRPES) to the C60/TiO2 (110) heterojunction in order to elucidate how the excited carriers behave at the junction. An experiment of TRPES utilizing the pump-probe method has been proven to be a powerful tool to simultaneously determine the electronic structure and the dynamics of the photoexcited carriers at semiconductor surfaces.
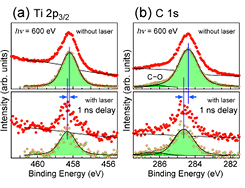
Fig. 1. (a) Ti 2p3/2 and (b) C 1s core-level spectra for C60/TiO2(110) with the C60 thickness of 0.57 nm. Spectra in the upper panels were acquired without the pump laser, whereas those in the lower panels were measured at 1 ns after irradiation of the laser pulse. The photon energy of the pump laser was 3.06 eV, and a power density was 40μJ/cm2/pulse. The photon energy of the probe SR light was 600 eV, and the emitted photoelectrons were collected with repetition rates of 208 kHz for the upper spectra and 1 kHz for the lower spectra. In each panel, a raw spectrum and a background-subtracted spectrum are drawn by dots. Background curves, shown by solid lines, were a combination of a Shirley-type curve and a linear line. Solid lines with shaded areas are the result of least-square fitting using Voigt functions.
The TRPES measurements were carried out at SPring-8 BL07LSU [1]. For the pump light, a second harmonic of an amplified Ti:sapphire laser pulse was used. The pulse duration and a repetition rate were 35 fs and 1 kHz, respectively. The photon energy was set at 3.06 eV, which exceeds both the band gap of rutile TiO2 (3.0 eV) and the energy difference between the highest occupied molecular orbital (HOMO) level and the lowest unoccupied molecular orbital (LUMO) level of C60 (2.3 eV). For the probe light, synchrotron radiation (SR) lights with the energies of 253 eV and 600 eV were used to measure valence-band spectra and Ti 2p and C 1s core-level spectra, respectively. The SR lights were provided by a single bunch and a 11/29-filling bunch train of an H-mode operation. SR pulses from the single bunches were used for the pump/probe measurements. A width of the pulse was 50 ps and a time interval between the pulses was 4.79μs.
We examined the effect of UV laser irradiation on the electronic structure of the C60/TiO2(110) heterojunction[2]. An upper panel in Fig. 1 shows a Ti 2p3/2 spectrum measured without UV irradiation. A background subtracted peak is reproduced by a single Voigt function with a peak maximum at 458.2 eV. A Ti 2p3/2 spectrum in the lower panel is acquired at 1ns after laser pulse irradiation. The peak is also fitted by a Voigt function with a peak maximum at 458.3 eV. Regarding the C 1s core-level peak of C60, a laser induced shift is 0.25 eV towards the higher binding energy (BE) side. The C 1s peak in the dark condition (without laser irradiation) is located at 284.4 eV. A small structure at 286.1 eV is due to C–O. The peak moves to 284.65 eV at 1 ns after irradiation. The BE shifts of both Ti 2p3/2 and C 1s peaks are transient because the peaks go back to the original positions by the time the next probe pulse arrives after 4.79 μs. The energy shift does not due to the surface photovoltage (SPV) effect since direction of the energy shift is opposite to the expectation. Moreover, the power density of the irradiated pump laser in the present study was 40μJ/cm2/pulse, which was insufficient to induce the SPV effect on the TiO2(110) surface [1].
Figure 1 clearly demonstrates that deposited C60 on TiO2(110) must be responsible for the UV-induced shift of the Ti 2p3/2 core level in the weak excitation condition. Aside from the TiO2 substrate, the C60 overlayer also absorbs the 3.06-eV pump laser. In the present excitation condition, the HOMO→LUMO+1 and HOMO-1→LUMO transitions are possible so that the valence electrons should promote to the LUMO and LUMO+1 levels, and the holes are left in the HOMO and HOMO-1 levels. Since both the LUMO and LUMO+1 states energetically overlap to the conduction band of TiO2, a resonant transfer of the excited electrons is possible from C60 to TiO2. Such an interface charge transfer competes with the electron-hole recombination process. It is speculated that a large fraction of the photoexcited electrons in the C60 layer is possible to be transferred to TiO2. The transferred electrons, then, undergo deexcitation to the bottom of the conduction band of TiO2. As a result, C60 is cationized after laser irradiation. The C 1s peak shift towards the higher BE side must, therefore, be caused by this charging effect. Regarding the TiO2 side of the heterojunction, additional electrons are injected from the C60 layer so that the carrier density is increased at the TiO2 surface. The BE shift of the Ti 2p3/2 peak should, therefore, reflect the enlarged magnitude of downward band bending.
References
- [1] M. Ogawa et al., Rev. Sci. Instrum. 83, 023109 (2012).
- [2] K. Ozawa et al., Organic Electronics 31, 98 (2016).