
Solid-Solid Interconversion Coupled by Jahn-Teller-Like Distortion and Bond-Order for the Hydrogen-Bonded Molecular Conductor β'-[H3(Cat-EDO-TTF)2]BF4
Mori Group
In the field of molecular functional materials, “π-electronic properties” of (super)conductors and anti/ferro-magnets and “protonic properties” of anti/ferro-electrics and proton conductor have been investigated separately and independently. Our aim is to couple “π-electronic properties” and “protonic properties” and to create novel “proton and π-electron coupled (PEC) properties and functionalities”. Recently, in the catechol-fused tetrathiafulvalene (TTF) system [1], the metallic state of purely organic single-unit crystal of κ-H3(Cat-EDT-ST)2 [2], the quantum spin liquid state of κ-H3(Cat-EDT-TTF)2 [3], and electronic switching induced by deuterium and charge transfer of κ-D3(Cat-EDT-TTF)2 and κ-D3(Cat-EDT-ST)2 [4,5] have been unveiled. Here we report unprecedented solid-solid interconversion coupled by Jahn-Teller-like distortion of hydrogen-bond (H-bond) unit and bond-order for the molecular conductor β'-[H3(Cat-EDO-TTF)2]BF4 [6].
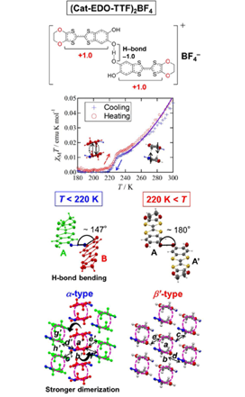
Fig. 1. The solid-solid interconversion from β’- (220 K < T) to α-[H3(Cat-EDO-TTF)2]BF4 (T < 220 K) is originated from the interplay of geometric Jahn-Teller-like distortion (from 180˚ to 147˚) of the intra-hydrogen-bond-unit [H3(Cat-EDO-TTF)2]¯ and magnetic stabilization of Cat-EDO-TTF dimers, where the paramagnetic-to-non-magnetic and low-to-high resistivity transition occurs. The solid line in the temperature dependence of χMT is reproduced by the singlet–triplet (S–T) dimer model with a very strong antiferromagnetic exchange coupling of 2J/kB = -1700 K.
The present crystal is composed of an H-bonded unit structure [H3(Cat-EDO-TTF)2]+ and an additional BF4¯ anion (positionally disordered), to give a chemical formula of [H3(Cat-EDO-TTF)2]BF4. In addition, this H-bond unit, [H3(Cat-EDO-TTF)2]+, is highly planar, and the H-bond part has an anionic symmetric [O..H..O]¯ structure with a short O..O distance (2.455(3) Å), whose H-bonded hydrogen occupies the inversion center of the H-bonded unit. Thus, two Cat-EDO-TTF skeletons in the unit are crystallographically equivalent to each other (Fig. 1). In the present EDO-TTF is +1 state, as estimated from empirical bond length analysis of the TTF skeleton due to the inclusion of the BF4¯ anion in the present system to maintain the charge neutrality of the overall crystal. As a result, the EDO-TFF system is expected to have a larger charge polarization within the H-bonded unit, [(EDO-TTF+-Cat)-[O..H..O]¯-(Cat-TTF+-EDO)]+ The present planar H-bonded molecular units form a sheet-like structure including the BF4¯ anions, which is further stacked to construct a so-called β’-type two-dimensional conducting layer composed of Cat-EDOTTF+ molecules. Thus, the high temperature phase (HTP: high temperature phase, 220 K < T) crystal can be called β’-[H3(Cat-EDO-TTF)2]BF4. As shown by red dashed circles in Fig. 1, the Cat-EDO-TTF+ molecules are π-dimerized in a head-to-tail manner with an interplanar distance of 3.41 Å, which is much shorter than that between the π-dimers (3.60 Å). Actually, the calculated transfer integral within the π-dimer (134 meV, denoted by a in Fig. 1) is much larger than that between the π-dimers (4.3 meV, denoted by b), suggesting that a very strong dimerization occurs. This type of conductive molecular material composed of a mono-oxidized TTF+ derivative is very rare due to the strong intermolecular Coulomb repulsion. In the present system, however, the head-to-tail arrangement of the significantly charge-polarized or zwitterionic-like Cat-EDO-TTF skeletons within and between the π-dimer(s) seems to effectively decrease the intermolecular Coulomb repulsions, to allow the construction of the infinite π–π stacking. Also, between the columns, there are some effective intermolecular interactions (c–e in Fig. 1).
The phase transition occurs around 220 K. Interestingly, upon the transition, the H-bonded unit becomes bent from 180° to 147° at one of the H-bonded oxygen atoms in the H-bond (Fig. 1), which results in desymmetrization of the H-bonded unit: two crystallographically equivalent Cat-EDO-TTF+ skeletons in the HTP (A and A’ molecules) become non-equivalent (A and B molecules), and, simultaneously, the symmetric [O..H..O]‒ H-bond is also desymmetrized to an [O–H..O]‒ fashion with the O–H and O..H distances of 1.17(4) Å and 1.31(4) Å, respectively. The HOMO energy levels of TTF skeletons are stabilized from -9.310 eV (HTP) to -9.322 and -9.335 eV (LTP: low temperature phase, T < 220 K) by extended Hückel calculation based upon crystal structure analyses. The bent of H-bond unit and stabilization of total energy from the HTP to the LTP are noted to be the Jahn-Teller-like distortion of H-bond unit. Curiously, however, the valence on the TTF skeletons in the LTP is fundamentally unchanged from that in the HTP, to maintain the +1 state, as revealed by the bond length analysis and DFT calculation. Also, the BF4 anions are still positionally disordered, however, notably the molecular arrangement of the Cat-EDO-TTF+ skeletons in the conducting layer is dramatically changed: the β’-type arrangement in the HTP, composed of a single kind of π-stack inclined in the same direction, transforms to a so-called α-type molecular arrangement in the LTP, where two types of the π-stacking columns with opposite inclinations (columns A and B) are alternately arranged. In this α-[H3(Cat-EDO-TTF)2]BF4 crystal, the tilt angle between the adjacent Cat-EDO-TTF+ skeletons in the columns A and B corresponds to the bend angle of the H-bonded unit, which thus suggests that this unusual molecular arrangement change is based on the transformation of the H-bonded unit between the planar and bent forms.
Temperature dependence of the electrical resistivity and magnetic susceptibility (Fig. 1) of this H-bonded-unit-based conductor [H3(Cat-EDO-TTF)2]BF4 was measured on the single crystal and polycrystalline sample, respectively. In both the measurements, we observed the phase interconversion between the high-temperature β’-[H3(Cat-EDO-TTF)2]BF4 and the low-temperature α-[H3(Cat-EDO-TTF)2]BF4, in the almost same temperature region. In the resistivity measurement, the resistivity at room temperature is 50 ohm cm and a semiconductor–semiconductor transition with an abrupt change in ρ was observed at around 210 K, which leads to an increase in the activation energy from 135 meV in the HTP to 165 meV in the LTP. This lowering of the transport property is due to the enhancement of the π-dimerization and the decrease of the inter-columnar interaction, which make the conductive π-electrons further localized within the π-dimer. The magnetic properties are also significantly altered around the anomaly at 220–230 K (Fig. 1): after its gradual decrease from 300 K, the χMT value rapidly dropped at 230 K and recorded 0.0 emu mol-1 K below 220 K, indicating that the LTP is a non-magnetic state. Also this temperature dependence was reversibly observed. We note here that the χMT value at 300 K (0.047 emu mol-1 K) is much smaller than the expected value for non-interacting two S = 1/2 spins (0.75 emu mol-1 K), suggesting the occurrence of a strong antiferromagnetic interaction between the S = 1/2 radical cationic TTF+ skeletons in the π-dimer. Actually, the temperature dependence of χMT in the HTP (T > 220 K) was well reproduced by the singlet–triplet (S–T) dimer model with a very strong antiferromagnetic exchange coupling of 2J/kB = -1700 K (solid line in Fig. 1).
In summary, we have discovered the novel proton-π-electron coupled (PEC) conductor, β’-[H3(Cat-EDO-TTF)2]BF4, with the solid-solid interconversion from β’- to α-phase accompanying resistivity and magnetic switching of the BF4 complex. This transition is derived from geometric Jahn-Teller-like distortion from planar to bent form of the composed hydrogen-bonded unit and the magnetic stabilization towards bond-order derived from spin singlet state with stronger dimerization of molecules. Further systematic tuning of proton and π-electron dynamics in this coupled system will afford novel chemical and physical functionalities beyond the framework of π-electronics so far.
References
- [1] H. Kamo, A. Ueda, T. Isono, K. Takahashi, and H. Mori, Tetrahedron Lett. 53, 4385 (2012).
- [2] T. Isono, H. Kamo, A. Ueda, K. Takahashi, A. Nakao, R. Kumai, H. Nakao, K. Kobayashi, Y. Murakami, and H. Mori, Nature Commun. 4, 1344 (2013).
- [3] T. Isono, H. Kamo, A. Ueda, K. Takahashi, M. Kimata, H. Tajima, S. Tsuchiya, T. Terashima, S. Uji, and H. Mori, Phys. Rev. Lett. 112, 177201 (2014).
- [4] A. Ueda, S. Yamada, T. Isono, H. Kamo, A. Nakao, R. Kumai, H. Nakao, Y. Murakami, K. Yamamoto, Y. Nishio, and H. Mori, J. Am. Chem. Soc. 136 (34), 12184 (2014).
- [5] A. Ueda, A. Hatakeyama, M. Enomoto, R. Kumai, Y. Murakami, and H. Mori, Chem. Eur. J. 21, 15020 (2015).
- [6] J. Yoshida, A. Ueda, A. Nakao, R. Kumai, H. Nakao, Y. Murakami, and H. Mori, Chem. Commun. 55, 15557 (2014).