
A Proton-Electron Coupled Molecular Conductor: Charge-Order Driven Proton Arrangement
Mori Group
There has been much attention on proton dynamics in a hydrogen-bond (H-bond) in a wide range of scientific fields from chemistry and physics to biology and materials science [1]. For example, in the quinhydrone complex, the proton transfer through an intermolecular H-bond associated with the electron transfer between the π-electronic systems gives rise to a unique phase transition in the single crystal under high hydrostatic pressure. Recently, H-bonded supramolecules demonstrate to be organic ferroelectrics, where proton dynamics are deeply related to π-electron based molecular orbitals. However, proton-electron coupled phenomena in the H-bonded molecular conductors have been little explored to date.
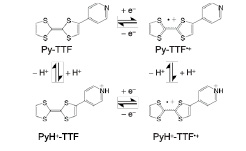
Fig. 1. A proton and an electron transfer process in Py–TTF.
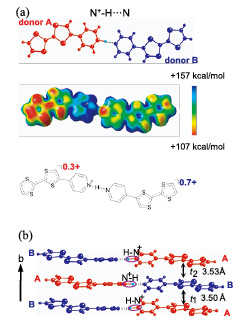
Fig. 2. (a) Structure of the H-bonded Py-TTF dimer by the X-ray crystal structure analysis and its electrostatic potential surface calculated by Gaussian 03 at the UB3LYP/6-311G(d,p) level of theory and (b) donor and proton arrangements in the CT complex along the b-axis, where the proton-electron coupled state as “charge-order driven proton arrangement” is realized.
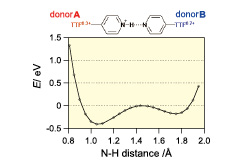
Fig. 3. N+-H distance dependence of the energy potential difference ΔE of the H-bonded dimer calculated by the UB3LYP/6-31G(d) level of theory. The initial molecular geometry (N+-H = 0.82 Å) was taken from the crystal structure, and the ΔE at 1.44 Å is set to 0 eV. The asymmetric shape where the minimum energy level (1.05 Å) is close to donor A with the charge-poor TTF0.3+ moiety suggests that the Coulomb repulsion between the positive charges of the TTF moiety and the proton is much smaller at the charge-poor donor A side.
In order to construct proton-electron coupled system in molecular conductors, we have focused on 4-pyridyl-substituted TTF derivative (PyTTF), where a strong electron donating TTF is directly bonded to a pyridyl substituent with both proton accepting (H-bonding) and electron withdrawing abilities (Figure 1). In this study, we have synthesized the proton-electron coupled CT complex based on PyTTF and an organic acceptor, tetrafluorotetracyanoquinodimetane (F4TCNQ). The complex is formulated as (PyH+–TTF0.3+Py–TTF0.7+)(F4TCNQ•¯)2•MeCN (1), containing the dimer structure where PyTTFs are jointed by H-bond (Figure 2). Detailed X-ray analysis by using synchrotron radiation reveals that proton arrangement of 1 is determined by the charge pattern of donor molecules, and we termed this state as “charge-order driven proton arrangement.” In this paper, we report the crystal structure and the electrical conductivity of 1.
The synchrotron X-ray diffraction was employed to experimentally determine the crystal structure including the position of the protons. In the crystals, there are five crystallographically independent molecules: two donors, two acceptors and one acetonitrile molecule as a crystal solvent. The donor molecules are connected by N+–H•••N type H-bond. The intra– and intermolecular distances N+–H and N-N are 0.82(5) Å and 2.781(3) Å, respectively. In the IR spectrum of 1, a broad peak was observed in 1800 – 2800 cm–1. This corresponds to the NH stretching absorption of N+–H•••N type H-bond.
In order to reveal the oxidation state of TTF moieties of donors A and B, we examined the charges for each molecule. In the IR spectrum, a lower wavenumber shift in the CN stretching mode was observed meaning that the charges of both acceptors C and D are –1. Since the net charges of the acceptors are –2, the net charge of donors A and B including the proton is estimated to be +2. The TTF moieties of the donors A and B are nearly planar and the dihedral angles between the TTF moiety and the pyridyl group of donors A and B are 15.14(6)° and 14.99(6)°, respectively. The central C=C bonds of the TTF moieties for donors A and B are 1.357(3) Å and 1.380(3) Å, respectively. These values are intermediate between neutral Py–TTF [1.344(9) Å] and cation radical salt of TTF [1.393 Å] indicating that donors A and B are partially oxidized. Each charge of TTF moieties of donors A and B was estimated to be +0.3 and +0.7 from their bond lengths. This estimation revealed that TTF moieties of donors A and B have different oxidation states and indicates the charge disproportionaion in the H-bonding unit. This charge disproportionation was also confirmed by the unrestricted density functional theory (DFT) calculation with the basis set 6-311G(d, p). These charge estimations provided the composition of 1 involving the charges of each molecules: (PyH+–TTF0.3+Py–TTF0.7+)(F4TCNQ•—)2•MeCN.
To clarify the reason why the proton is located close to the donor A in the H-bond, the potential curve of proton was calculated by the unrestricted DFT calculation (Figure 3). It is known that an N+–H•••N type H-bond exhibits a double–well potential curve of proton coordinates when an N–N distance is more than 2.5 Å. The calculated curve shows a double–well potential predicted from the N-N distance of 1 [2.781(3) Å]. The curve exhibits an asymmetric shape in which the charge disproportionation in H-bonding unit is reflected and takes a minimum energy level close to donor A containing charge-poor TTF0.3+ moiety. This is due to suppress the intramolecular Coulomb repulsion between TTF moiety and pyridinium group. It is likely that this effect causes the proton ordering, suggesting a proton-electron coupled state in 1.
Finally, we investigated the effect of the charge ordering on the conductivity. The complex exhibits semiconducting behavior with room temperature resistivity of 44 ohm cm and activation energy of 0.12 eV. The complexes with 1:1 stoichiometry of donor to F4TCNQ ratio have a tendency to show high resistivity around 104 ohm cm because the strong electron accepting ability of F4TCNQ causes a fully–reduced state in CT salts. However, the complex 1 showed a resitivity less than typical 1:1 F4TCNQ salts by two orders of magnitude. This indicates that the relatively high conductivity of 1 is originated from charge-ordered donor column. In addition, this results support the existence of proton because if proton didn’t exist, fully-oxidized TTF would form a completely-filled band structure. Moreover, the resistivity is tremendously suppressed by pressure under 1.8 GPa by three orders of magnitude at room temperature, indicating the suppression of the charge ordering. The behaviors of protons coupled with melting of the charge ordering under pressure is the next research target.
In summary, we have successfully prepared the protonated CT complex based on pyridyl-substituted TTF derivative. The asymmetrical double-well proton potential in which the charge disproportionation is reflected explained the reason why a proton in H-bond is ordered. Proton arrangement in donor columns is determined by the pattern of charge ordering, and therefore we named this novel proton-electron coupled system as “charge-order driven proton arrangement”. In this system, we believe that proton dynamics coupled with a change of electronic state by external stimuli such as electric field, light, pressure, etc. is observed.
References
- [1] T. Mitani, G. Saito, and H. Urayama-Mori, Phys. Rev. Lett. 60, 2299 (1988); H. Suzuki, H. Mori, J. Yamaura, M. Matsuda, H. Tajima, and T. Mochida, Chem. Lett. 36, 402 (2007); H. Ohchi, K. Takahashi, J. Yamaura, S. Takaishi, and H. Mori, Physica B 405, S341 (2010).