
Development of Novel First-Principles Methods for Material Research
Sugino Group
The first-principles computational methods have enabled, with increasing precision, to elucidate properties of matters starting from the microscopic quantum principles only. This was strongly driven by the development of supercomputers and the computational theory, e.g., density functional theory (DFT) and Hartree-Fock. Those theories have already matured and great many efforts are now made toward the development of post-DFT theory and the extension of the frontier of DFT applications. Towards the goal, our group has made three important steps. The first one is the development of a post-DFT scheme, which is based on the tensor compression technology to compactly represent the many-body wave function. A graduate course student, Wataru Uemura, showed that the canonical decomposition algorithm indeed greatly facilitates handling of the wave function that appears in the full configuration interaction (CI) scheme. The developed scheme [1], called symmetric tensor decomposition (STD), is considered make the full CI a method of choice for the molecular and some of the condensed matter researches.
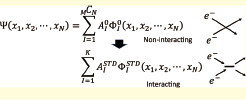
Fig.1: An electronic wave function has been traditionally expanded by non-interacting terms (Φ0) in the configuration interaction (above). The series can be greatly shortened when expanded by interacting ones (below). The tensor decomposition provides a practical way to construct a maximally compact series, allowing thereby accurate numerical handling of the wave function. Using this novel algorithm, or STD-CI, we are developing a code for electronic structure calculation to be used in the next-generation supercomputer.
The use of the tensor compression technology itself is not new, but was established as a powerful method to handle one-dimensional spin and Hubbard systems some years ago. The STD-CI, however, applied the technology to the conventional molecular orbital theory and, with algorithmic improvements, has greatly reduced the memory and the computational time requirement. It is also anticipated that, by combining STD-CI with the tensor network theory developed in the spin physics, extended systems will become the target of study. Despite the improved efficiency, the computational time is yet prohibitively large even for medium-sized molecules, but is expected to become tractable by the next-generation supercomputers because of its excellent parallelizability.
Towards the study of electronically excited states, the research associate, Yoshifumi Noguchi parallelized the code for many-body perturbation theory, or a post-DFT theory, for the K-computer and the ISSP supercomputer. The excited-states of a molecule containing up 100 atoms, such as fullerenes, has become the target of study [2]. By combining with the DFT-based schemes [3], we are advancing the method for excited-state research.
Our group regards the solid-liquid interface is the important application field of DFT. We are particularly interested in explaining the energy conversion mechanism as typified by the fuel-cell reactions. The major object of the fuel-cell science is to relate the bias potential with the electrocatalytic reactions, which has been hampered by the difficulty of applying the bias potential to the interface. Important algorithmic improvements were made by the collaboration with research group in AIST to keep applying the bias potential throughout the DFT-based molecular dynamic simulation [4]. Those methods are now used for the large-scale simulations on K-computer.
References
- [1] W. Uemura and O. Sugino, Phys. Rev. Lett. 109, 253001 (2012).
- [2] Y. Noguchi, O. Sugino et al., J. Chem. Phys. 137, 24306 (2012).
- [3] T. Tsukagosi et al., Phys. Rev. B 87, 35421 (2012); Phys. Rev. A 86, 64501 (2012).
- [4] N. Bonnet et al., Phys. Rev. Lett. 109, 266101 (2012).